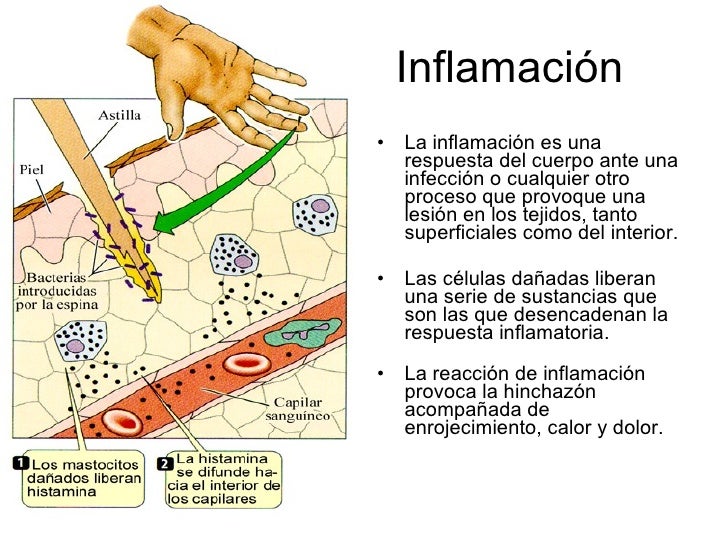
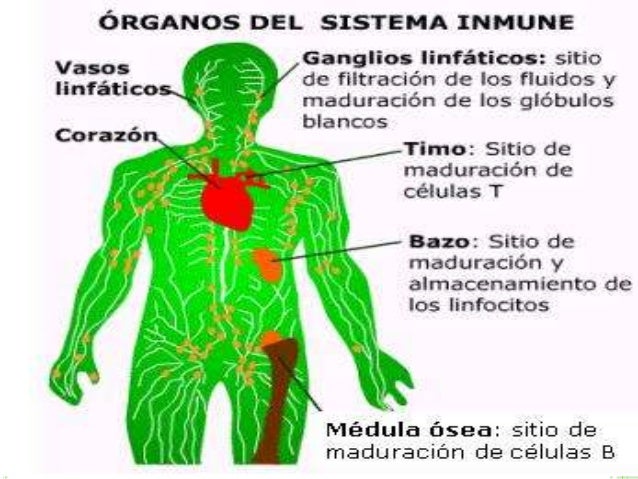
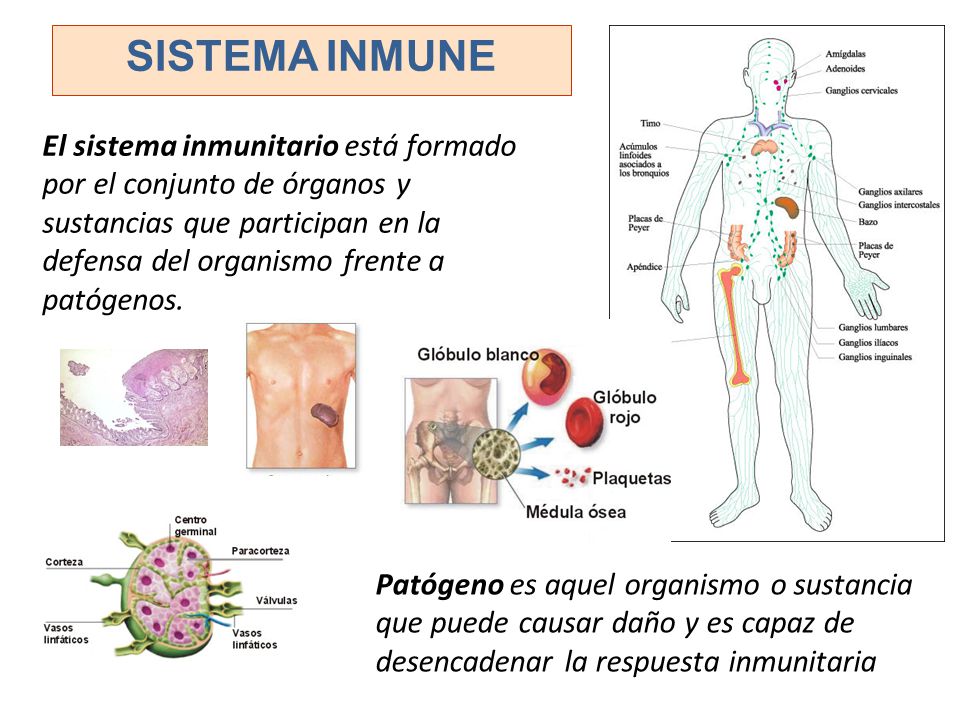
El sistema inmunológico es la defensa natural del cuerpo contra las infecciones, como las bacterias y los virus. A través de una reacción bien organizada, su cuerpo ataca y destruye los organismos infecciosos que lo invaden. Estos cuerpos extraños se llaman antígenos.
La inflamación es la respuesta del sistema inmunológico a los antígenos. Como respuesta a la infección o la lesión, diversas clases de glóbulos blancos se transportan por el torrente sanguíneo hasta el lugar de la infección y solicitan más glóbulos blancos. Cuando la amenaza desaparece, la inflamación cede. Por ejemplo, cuando una persona se corta o tiene gripe, la inflamación se usa para matar la bacteria o el virus que invade el cuerpo.
En las personas que gozan de buena salud, el sistema inmunológico puede distinguir entre los tejidos propios del cuerpo y los extraños que lo invaden, tales como virus y bacterias. En algunos tipos de artritis, como la artritis reumatoide, el sistema inmunológico no funciona correctamente. Cuando esto ocurre, el sistema inmunológico:
No identifica la diferencia entre los tejidos propios del cuerpo y los agentes que lo invaden tales como las bacterias y los virus.
Produce, por error, inflamación en contra de tejidos o partes del cuerpo normales, tales como las articulaciones, como si éstos fueran agentes extraños que lo invaden.
Se desconocen las razones por las que el sistema inmunológico no funciona correctamente.
Las enfermedades que se desarrollan cuando el sistema inmunológico no funciona correctamente se denominan enfermedades autoinmunes.
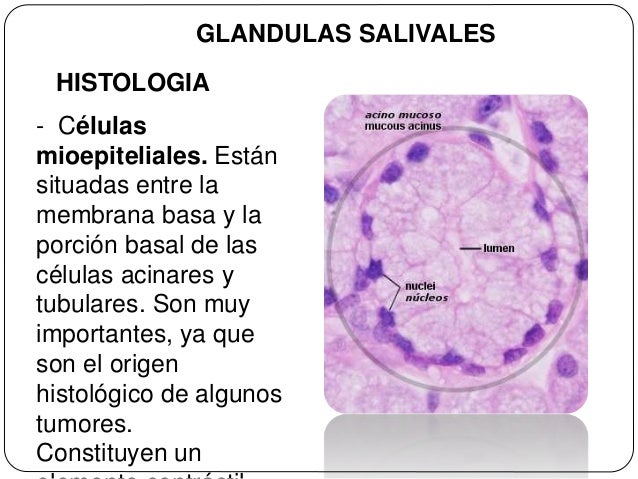
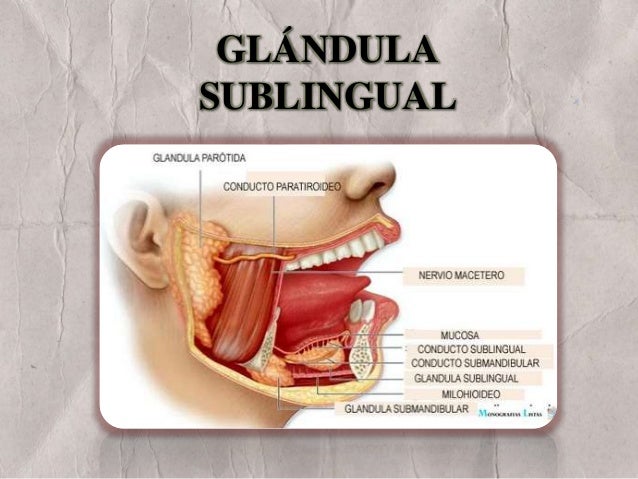
Las glándulas salivales en diversas especies biológicas son glándulas exocrinas en el sistema digestivosuperior que producen la saliva que vierten en la cavidad oral. La saliva es un líquido incoloro de consistencia acuosa o mucosa, que contiene proteínas, glucoproteínas, hidratos de carbono y electrólitos, células epiteliales descamadas y leucocitos. Su función, entre otras, es iniciar la digestión de los alimentos al humedecerlos para ayudar en el proceso de masticación y deglución, y contiene enzimas que comienzan el proceso de digestión de carbohidratos y grasas. A continuación te explicamos qué son y cómo funcionan las glándulas salivales.
Glándula parótida
La glándula salival más voluminosa es la parótida, ubicada por debajo del arco cigomático, por delante de la apófisis mastoides y detrás de la rama de la mandíbula. Se relaciona con las ramas principales del nervio facial. Dentro de su substancia asciende la arteria temporal superficial. La secreción de la glándula parótida es de tipo seroso (fluída).
El conducto parotídeo abandona el ángulo anterosuperior de la glándula salival, cruza sobre el músculo masétero, perfora el músculo buccinador, y se abre en el vestíbulo bucal, superior frente al segundo molar superior.
Glándula submandibular
La glándula submandibular de la cavidad bucal produce una secreción salival mixta, serosa y mucosa (viscosa) pero predominantemente seroso. Está por dentro del ángulo mandibular. Su conducto pasa hacia adelante y adentro, en el piso de la boca, y se abre al lado del frenillo de la lengua.
Glándula sublingual
La sublingual de la boca es la más pequeña de las glándulas salivales. Esta glándula, predominantemente mucosa, está por debajo de la mucosa del piso de la boca. Su secreción salival fluye a través de varios conductos sublinguales separados que se abren en el pliegue sublingual.
Si deseas leer más artículos parecidos a Las glándulas salivales - Función y qué son, te recomendamos que entres en nuestra categoría de Formación.
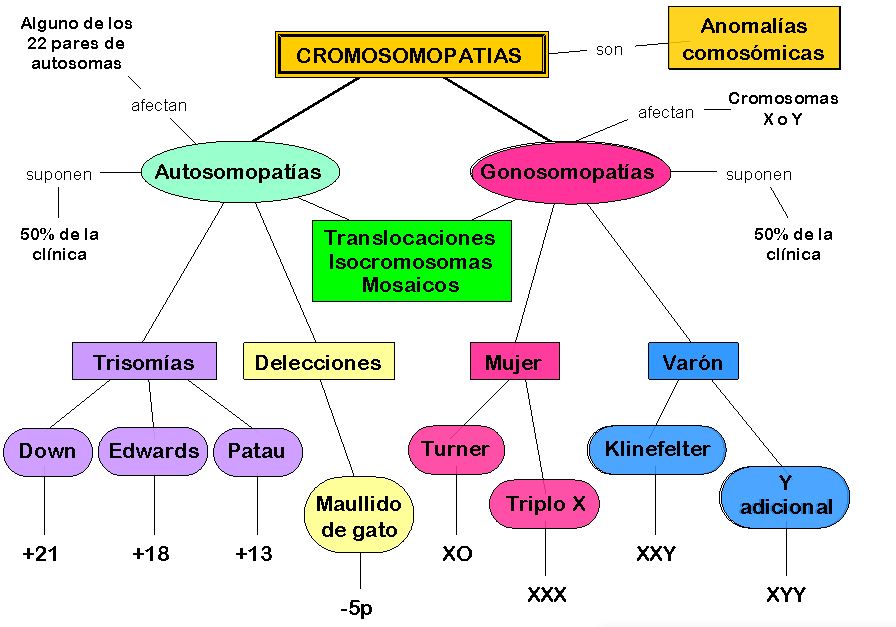
Estas enfermedades se deben a alteraciones en la estructura de los cromosomas, como pérdida o deleción cromosómica y encontramos las siguientes:
Síndrome de Down o trisomía 21:
Debida a la presencia de una copia adicional de la zona crítica en 21q22.1, que puede deberse a una trisomía 21 en más del 94% de los casos, una translocación con otros cromosomas (heredado o no) en el 3.3%, la coexistencia de una línea celular con la trisomía 21, otras, lo que se denomina mosaicismo en el 2.4%.
Se asocia a una edad materna avanzada. Presentan hipotonía, talla baja, braquicefalia, facie aplanada, epicanto, orejas pequeñas con hélix plegado y protrusión lingual. Tienen exceso de piel en la nuca, cardiopatía congénita en el 50%, malformaciones gastrointestinales, pueden hacer reacciones leucemoides y neoplasias hematológicas. Debe descartarse hipotiroidismo. Las manos son cortas y anchas, presentan clinodactilia de meñiques y surco palmar transverso, en pies hay surco plantar tibial, e hipermovilidad articular.
Síndrome de Edwards o trisomía 18:
Debida a la presencia de tres cromosomas 18. También hay una correlación con edad materna avanzada. El pronóstico de sobrevida es muy pobre. Los rasgos clínicos incluyen retraso severo de crecimiento, hipertonía, occipucio prominente, blefarofimosis, microstomía, micrognatia, orejas pequeñas, esternón corto, cardiopatías severas, sobreposición de dedos 2° y 5° sobre 3° y 4°, pie en balancín, uñas hipoplásicas, malformaciones renales y criptorquidia.
Síndrome de Patau o trisomía 13:
Debida a la presencia de tres cromosomas 13. Se relaciona con edad materna avanzada, es clínicamente muy severa y generalmente letal antes de los tres meses de edad. El fenotipo incluye malformaciones del sistema nervioso central, hipotelorismo ocular, microftalmia, fisura labiopalatina, polidactilia postaxial, malformaciones cardiacas y renales, agenesia de cuero cabelludo, hemangiomas, criptorquidia y útero bicorne.
Síndrome de Wolf Hirschhorn o 4p-:
Debida a la deleción parcial de un segmento del brazo corto del cromosoma 4 que involucre 4p16.3. Presentan retraso severo de crecimiento con microcefalia. Tienen un perfil característico con el puente nasal alto, nariz ganchuda, filtrum corto, micrognatia, comisuras orales hacia abajo, fisura labiopalatina y orejas de implantación baja. Coloboma del iris, hipertelorismo e inclinación antimongoloide de los ojos. Defecto septal ventricular, isomerismo pulmonar, hernia diafragmática, mesenterio común, hipoplasia renal, hipospadias y retraso de la edad ósea.
Síndrome de Cri du Chat o 5p-: debida a la deleción parcial de un segmento del brazo corto del cromosoma 5 que involucre 5p15.2. Aunque la mayoría de las deleciones ocurren como mutaciones de novo, en alrededor de un 12% resultan de una translocación presente en uno de los padres. Microcefalia, fascie redonda, hipertelorismo ocular, epicanto, estrabismo, fisuras palpebrales hacia abajo, micrognatia, orejas de implantación baja, cardiopatía congénita, e hipotonía, con severo retardo psicomotor. En recién nacidos el llanto agudo similar al maullido de un gato es muy sugerente. Pueden vivir hasta la adultez.
Síndrome de Turner o monosomía X:
Presencia de un sólo cromosoma X, también pueden verse diversas alteraciones estructurales en un cromosoma X y no es rara la presencia de dos o más líneas celulares diferentes. Talla baja, disgenesia gonadal, cuello alado, linfedema del dorso de pies y manos, uñas angostas, cúbito valgo, paladar ojival, tórax ancho, malformaciones renales y cardiovasculares. Tienen mayor riesgo de desarrollar otitis media, hipertensión, enfermedad tiroidea y diabetes. Hay un riesgo aumentado de algunos trastornos en el desarrollo de lenguaje, conducta neuromotora y aprendizaje. Infertilidad.
Síndrome Klinefelter o 47,XXY:
presencia de un cromosoma X adicional en un varón. Los rasgos clínicos en niños son mínimos y el diagnóstico puede retrasarse hasta la adolescencia, por talla alta, trastornos de aprendizaje y testículos pequeños. Infertilidad.
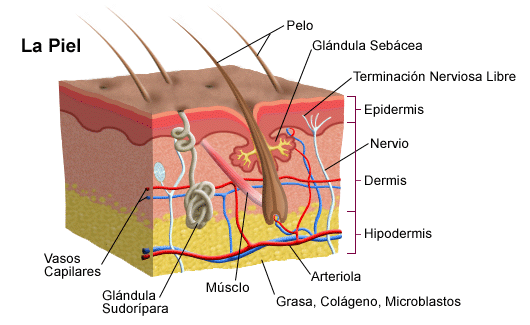
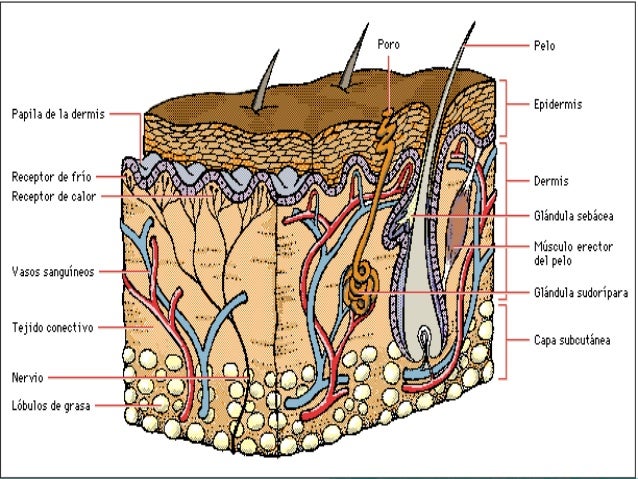
La piel es el órgano más grande del cuerpo y lo cubre completamente. Además de servir como protección contra el calor, la luz, las lesiones y las infecciones, la piel también:
Regula la temperatura del cuerpo
Almacena agua y grasa
Es un órgano sensorial
Impide la pérdida de agua
Impide el ingreso de bacterias
A lo largo de todo el cuerpo, las características de la piel varían (por ejemplo, su grosor, color y textura). Por ejemplo, la cabeza contiene más folículos capilares que cualquier otro lugar, mientras que las plantas de los pies no contienen ninguno. Además, las plantas de los pies y las palmas de las manos tienen una piel mucho más gruesa que otras áreas del cuerpo.
La piel está formada por las siguientes capas, y cada una de ellas con funciones específicas:
Epidermis
Dermis
Capa de grasa subcutánea
Epidermis
La epidermis es la capa externa delgada de la piel que consta de tres tipos de células:
Células escamosas. La capa más externa se pela continuamente.
Células basales. Las células basales se encuentran debajo de las células escamosas.
Melanocitos. Los melanocitos se encuentran en todas las capas de la epidermis y forman la melanina, que le da el color a la piel.
Dermis
La dermis es la capa intermedia de la piel. Contiene lo siguiente:
Vasos sanguíneos
Vasos linfáticos
Folículos capilares
Glándulas sudoríparas
Estructuras de colágeno
Fibroblastos
Nervios
La dermis se mantiene unida mediante una proteína llamada colágeno, que está formada por fibroblastos. Esta capa le da a la piel flexibilidad y fuerza. Además contiene receptores de color y tacto.
Capa de grasa subcutánea
La capa de grasa subcutánea es la capa más profunda de la piel y consta de una red de colágeno y células de grasa. Ayuda a conservar el calor del cuerpo y protege el cuerpo de lesiones al actuar como absorbedor de golpes.
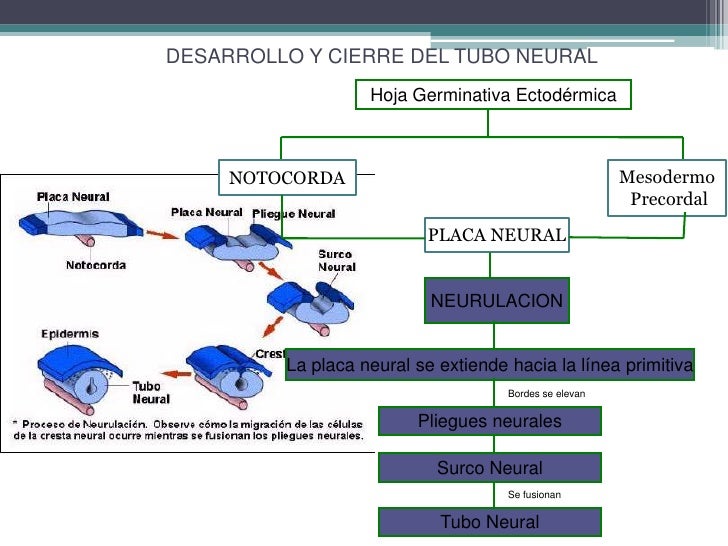
a neurulación es el proceso por el cual se forma el tubo neural durante el desarrollo intrauterino. El tubo neural resulta fundamental para la diferenciación de las células del sistema nervioso central, mientras que las crestas neurales, estructuras asociadas a la que nos ocupa, lo son para la formación del sistema nervioso periférico.
En este artículo describiremos las dos fases de la neurulación o formación del tubo neural: la primaria, en la que la placa neural empieza a replegarse sobre ella misma, y la secundaria, que culmina este proceso y permite el desarrollo posterior del sistema nervioso.
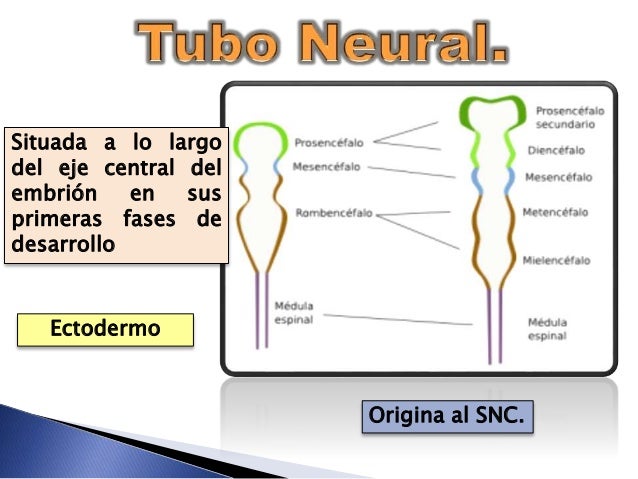
Tubo neural
El tubo neural es una estructura embrionaria que se forma durante el primer mes de la gestación; en concreto, el tubo acaba de cerrarse alrededor de la semana 28 después de la fecundación. Se trata del precursor del sistema nervioso central, compuesto por el encéfalo y la médula espinal.
A medida que el desarrollo embrionario progresa el tubo neural se divide en cuatro secciones: el encéfalo anterior (prosencéfalo), el medio (mesencéfalo), el posterior (rombencéfalo) y la médula espinal. Cada una de estas partes progresará hasta dar lugar a los diferentes elementos que componen el sistema nervioso central adulto.
Mientras que la mayor parte del sistema nervioso se desarrolla a partir de las paredes del tubo neural, también es relevante el hueco que se encuentra entre las paredes: el neurocele o canal neural. Esta estructura se transformará progresivamente en los ventrículos y el resto de cavidades del encéfalo, a través de los cuales circula el líquido cefalorraquídeo.
La neurulación primaria
Tras la fecundación se forma el cigoto, la célula primigenia compuesta por la fusión de un óvulo y un espermatozoide. El cigoto se divide sucesivamente, convirtiéndose en un conjunto de células que se denomina mórula. Posteriormente aparece el blastocele, una cavidad llena de fluido, dentro de esta estructura; cuando esto sucede hablamos de “blástula”.
Más adelante la blástula se divide en tres capas: el endodermo, el mesodermo y el ectodermo. Cada una de estas secciones dará lugar a distintas partes del organismo. El ectodermo es la más importante para el asunto que nos ocupa, puesto que a partir de éste se desarrolla el sistema nervioso, tanto el central como el periférico.
La notocorda, una estructura que se localiza en el mesodermo, envía señales a las células que se encuentran a su alrededor. Las que no reciben dichas señales se transforman en la placa neural o neuroectodermo, un conjunto de células que ya se han especializado en funciones nerviosas. La palabra “placa” hace referencia al aspecto aplanado del neuroectodermo.
La neurulación primaria consiste en la proliferación de células nerviosas en la placa neural. Estas hacen que la placa se transforme en el tubo neural, un paso fundamental en el desarrollo del organismo de los seres humanos.
Formación y cierre del tubo neural
Durante el proceso de neurulación la placa neural se aplana, se alarga y se pliega sobre sí misma en torno al surco neural, que acaba teniendo forma de U a medida que las paredes se levantan, formando las crestas neurales y el tubo neural. En este momento del proceso el tubo está abierto por ambos extremos; nos referimos a los neuroporos caudal y rostral.
Lo normal es que estas aperturas se cierren después de unos días; sin embargo, en ocasiones el tubo no se cierra correctamente, lo cual da lugar a trastornos como la espina bífida (que afecta a la columna vertebral) y la anencefalia (asociada a malformaciones muy graves en el cerebro).
Es importante diferenciar el tubo neural de la cresta neural debido a que el primero se transforma en la mayoría de estructuras del sistema nervioso central, mientras que el periférico es una progresión de la cresta neural.
La neurulación secundaria
La neurulación secundaria es el proceso que culmina la formación del tubo neural. Éste no se debe a las señales enviadas por determinadas células, como sucede con la neurulación primaria, sino que se da como consecuencia del propio desarrollo del tubo neural.
Este proceso se asocia con la división de las células del tubo neural entre mesenquimatosas y epiteliales. Las primeras se localizan en la parte central del tubo, y las segundas en su región periférica. A medida que estas células se diferencian se forman cavidades entre los dos conjuntos.
Las células mesenquimatosas que se localizan en esta parte del embrión se condensan y forman lo que conocemos como cordón medular; éste, a su vez, se ahueca por dentro hasta dejar paso a la cavidad del tubo neural. Este fenómeno se inicia en la región sacra de la columna vertebral.
Así pues, mientras que la neurulación primaria consiste en el repliegue de la placa neural sobre sí misma, la secundaria se corresponde con el vaciado de la cavidad del tubo neural, muy asociada a la diferenciación de las células del sistema nervioso del embrión.
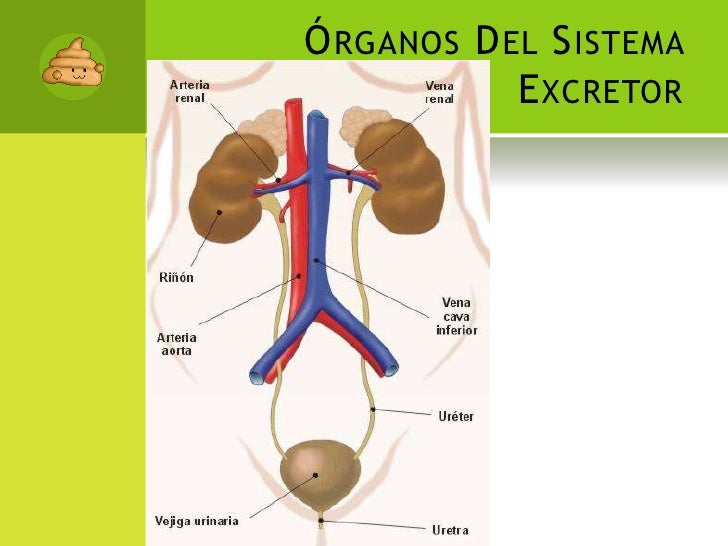
Primero que todo es importante tener claro qué es el sistema urinario y por qué órganos está compuesto. El sistema urinario es un conjunto de órganos que trabajan junto con el pulmón, la piel y el intestino excretando productos de desecho para mantener la homeostasis del organismo, el principal producto de desecho del sistema urinario es la urea, producto de la descomposición de alimentos proteicos en el cuerpo.
Uno de los órganos más importantes de este sistema es el riñón, es un órgano en forma de frijol, aproximadamente del tamaño de nuestro puño, es un órgano retroperitoneal y se localiza más exactamente debajo de la caja torácica. Los riñones eliminan urea a través de sus unidades funcionales, la nefrona, cada nefrona posee una bola de capilares sanguíneos llamada glomérulo y una serie de túbulos llamados túbulos renales, la orina se produce a medida que pasa por los glomérulos y los túbulos, cada parte del túbulo tiene una función diferente para reabsorción y excreción. De los riñones la orina viaja a la vejigapor dos tubos de aproximadamente 8 a 10 pulgadas de largo, muy delgados llamados uréteres, alrededor de cada 10 a 15 segundo se vacían pequeñas cantidades de orina desde los uréteres y hacia la vejiga.
La vejiga es un órgano muscular hueco en forma de globo que se encuentra ubicado sobre la pelvis, se encarga de almacenar la orina, puede llegar a almacenar hasta 16 onzas de orina de 2 a 5 horas, hasta que estemos listos para ir al baño a expulsarla.
El sistema urinario empieza a desarrollarse a partir de 3 estructuras principales.
·PRONEFROS: Formado por 7-10 grupos celulares que forman losnefrostomas, unidades vestigiales excretoras, al final de la cuarta semana el sistema pronéfrico desaparece dejando sus vestigios que son los nefrostomas.
·MESONEFROS: Mientras se da la regresión del sistema pronéfrico, se forman los primeros túbulos excretores del mesonefro que se alargan formando una S y dando origen a una serie de capilares que formaran un glomérulo y alrededor del glomérulo los túbulos forman la capsula de Bowman y estas dos estructuras juntas forman el corpúsculo renal. El túbulo se inserta lateralmente en el conducto colector longitudinal llamadoconducto mesonéfrico o conducto de Wolff. Los túbulos caudales todavía se están desarrollando mientras que los craneales se degeneran y desaparecen en el aparato urogenital femenino.
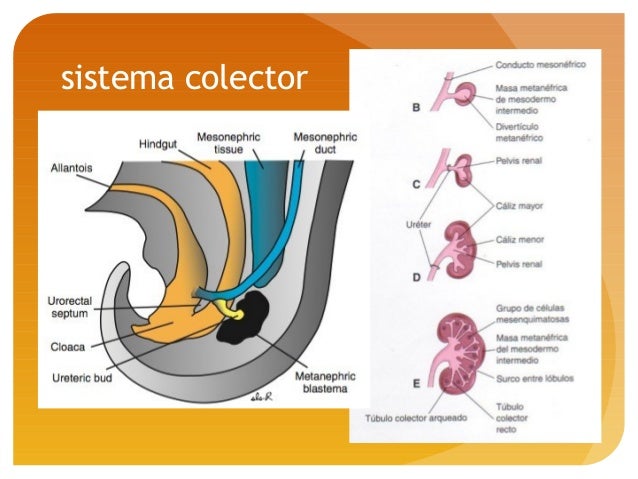
·METANEFROS: Estas unidades secretoras se desarrollan a partir delmesodermometanéfrico a partir de él se desarrolla el riñón definitivo y elsistema colector y el sistema excretor.
SISTEMA COLECTOR:
Los conductos excretores del riñón se desarrollan a partir de una excrecencia del conducto mesonéfrico cerca de su entrada a la cloaca, llamado yema uretral. Más adelante la yema se dilata formando la pelvis renal primitiva que se divide en una parte craneal y otro caudal que serán lo cálices renales mayores. A medida que penetran el tejido metanéfrico se siguen dividiendo hasta llegar más o menos a 12 túbulos y este desarrollo continúa hasta finales del quinto mes llegando a la formación de los cálices menores. Posteriormente los túbulos colectores se alargan y convergen en el cáliz menor, formando allí la pirámide renal.
SISTEMA EXCRETOR:
Cada túbulo colector formado está cubierto por un casquete de tejido metanéfricoen su parte distal, estas células del casquete forman pequeñas vesículas llamadasvesículas renales y estas a su vez generan unos túbulos en forma de S.
Los capilares crecen en un extremo de la S y se diferencian en glomérulos, ambos, túbulos y glomérulos forman las nefronas o unidades excretoras, que es la unidad funcional del riñón,y el extremo proximal de cada nefrona desarrolla la cápsula de Bowman, mientras que el extremo distal y su alargamiento continúo da lugar a eltúbulo contorneado proximal, el asa de Henle y el túbulo contorneado distal.
Las nefronas se desarrollan hasta el momento del nacimiento, donde tenemos cerca de 1 millón de nefronas por riñón, al momento del nacimiento los riñones tienen una forma lobulada que con el tiempo desaparece debido al crecimiento de las nefronas, pero cabe resaltar que en este crecimiento no se aumenta su número o cantidad, simplemente su tamaño.
POSICIÓN DEL RIÑÓN:
Al comienzo del desarrollo urogenital el riñón se empieza a formar en la región pélvica en una posición caudal, pero a medida que se desarrolla pasa a la región abdominal en una posición más craneal debido al crecimiento y perdida de la curvatura corporal del feto.
FUNCIÓN DEL RIÑÓN:
El riñón definitivo formado a partir del metanefros pasa a ser un funcional en la semana 12, la orina pasa a la cavidad amniótica y se mezcla con el líquido amniótico, durante el periodo fetal el riñón no realiza su función de excreción de productos de desecho debido a que esta función es realizada por la placenta.
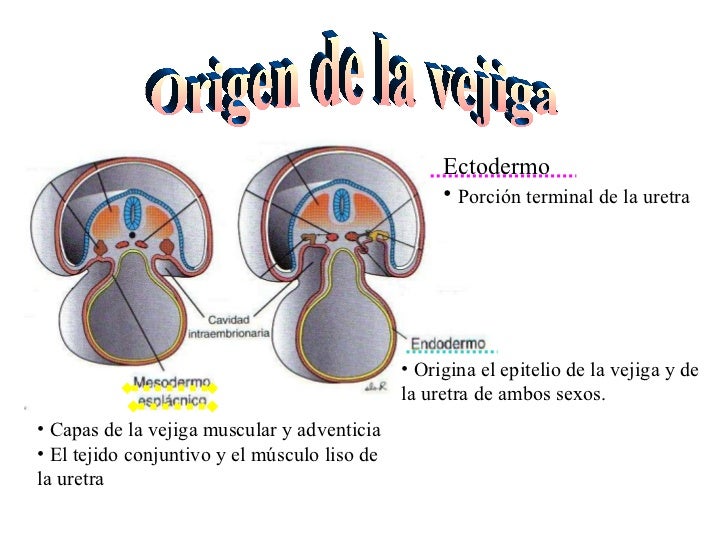
VEJIGA Y URETRA:
Aproximadamente de la 4ta a la 7ma semana la cloacase divide anteriormente en elseno urogenital y posteriormente en el conducto anal, el seno urogenital se diferencia en 3 partes, la superior y además más grande es la vejiga urinaria.
Al principio la vejiga es continua con el alantoides pero cuando la luz del alantoides desaparece, el uracose mantiene y se une al ápice de la vejiga y el ombligo que en el adulto es el ligamento umbilical medio. La siguiente parte del desarrollo origina la parte prostática y membranosa de la uretra, se origina es en el sexo masculino y es un conducto estrecho desarrollado a partir de la parte pélvica del seno urogenital.(imagen)
La última parte es la parte fálica del seno urogenital, esta aplanada en ambos extremos y a medida que el tubérculo genital crece, esta parte se extiende ventralmente, el desarrollo de la parte fálica del seno urogenital difiere ampliamente entre los dos sexos.
Durante la diferenciación de la cloaca, las partes caudales de los conductos mesonéfricos desaparecen absorbidos por las paredes de la vejiga y quedan como sus excrecencias los uréteres, los cuales entran a la vejiga por separado y como consecuencia del ascenso de los riñones los orificios uretrales se desplazan hacia una posición más craneal. Mientras tanto los orificios de los conductos mesonéfricos, en el sexo masculino se desplazan para introducirse en la uretra prostática para posteriormente pasar a ser los conductos eyaculadores.
Desarrollo del Aparato reproductor masculino y femenino
El desarrollo del aparato reproductor se divide en dos periodos, uno indiferenciado y otro diferenciado. El indiferenciado se caracteriza por la presencia en el embrión de primordios gonadales, gonoconductos, seno urogenital y genitales externos con características comunes para ambos sexos. En el diferenciado, estos primordios tendrán un desarrollo masculino o femenino según el sexo que se determinó en el momento de la fecundación. Es importante distinguir las características de cada uno de los períodos para entender la relación que debe existir entre: sexo génico, sexo cromosómico, sexo gonadal, sexo hormonal, sexo genital interno y externo, así como la edad gestacional en que se determina su diferenciación.
1) Período indiferenciado
Duración en varones y hembras
Características de los primordios de las gónadas, gonoconductos, seno urogenital y genitales externos:
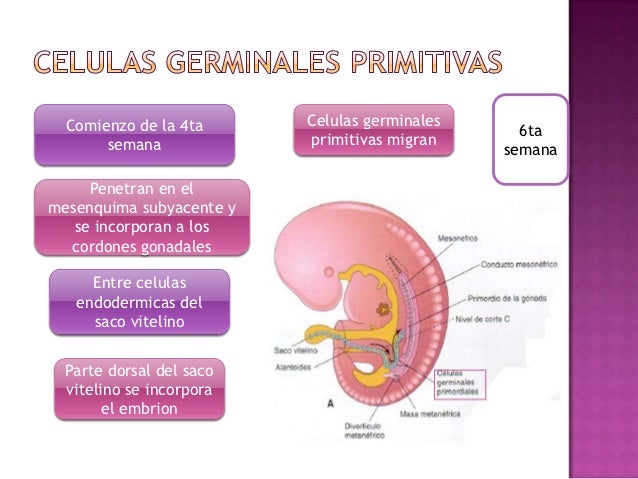
Cresta gonadal: constituyentes y hoja embrionaria de la que se origina. Importancia de las células germinativas primitivas: localización, migración y función en la diferenciación de la cresta gonadal.
Gonoconductos mesonéfricos y paramesonéfricos: edad de aparición, localización e identificación
Cloaca primitiva: división y formación del seno urogenital
Genitales externos: primordios, edad gestacional e identificación
2) Importancia del cromosoma Y en la diferenciación sexual. Gen SRY. Función e importancia.
3) Periodo diferenciado. Testículo y ovario. Edad gestacional, evolución, y derivados de:
Células germinativas primitivas
Cordones sexuales primarios y secundarios
Mesodermo intermedio
Corteza y médula gonadal
Epitelio celómico
4) Genitales internos masculinos y femeninos.
Importancia de la testosterona y del factor inhibidor del Müller (hormona anti-mülleriana) en el varón.
Evolución y derivados del conducto mesonéfrico o de Wolff
Evolución y derivados del conducto paramesonéfrico o de Müller
Seno urogenital. Derivados genitales y no genitales en la hembra y en el varón
Formación de vagina. Primordios. Bulbos sinovaginales.
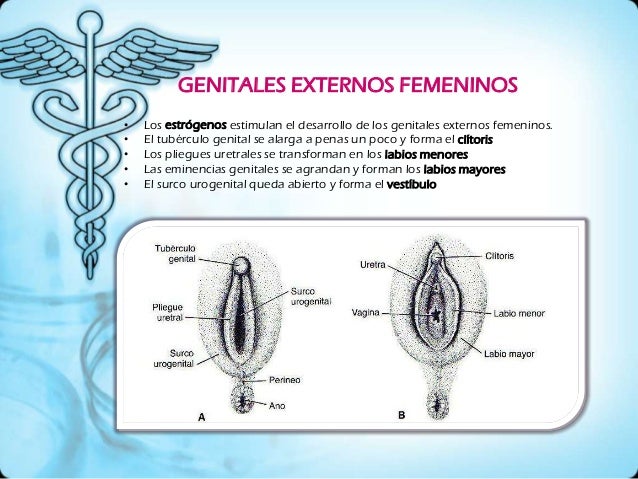
5) Genitales externos.
Derivados definitivos de los primordios femeninos y masculinos.
Importancia de la dihidrotestosterona en el varón
6) Descenso de las gónadas.
Testículo: importancia del ligamento genital caudal, el gubernáculo y del conducto peritoneo vaginal.
7) Anomalías congénitas del Aparato Reproductor
Alteraciones en el desarrollo de las gónadas.
Síndromes de Klinefelter y Turner. Características y causa.
Alteraciones en el descenso testicular.
Definición y mecanismo embriopatológico de:
Ectopia testicular
Criptorquidia
Hidrocele
Hernia inguinoescrotal
Anomalías müllerianas. Características y mecanismo embriopatológico de:
Agenesia uterina y de parte de vagina
Útero unicorne
Útero didelfo y bicorne
Útero arcuato y septado
Anomalías de genitales masculinos. Definición y mecanismo embriopatológico de:
Hipospadias
Epispadias y extrofia vesical
Alteraciones del desarrollo sexual
Sexo cromosómico, causas, características de las gónadas, genitales internos y externos de:
Hermafroditismo verdadero (Trastorno del desarrollo sexual ovotesticular)
Pseudohermafroditismo masculino (Trastorno del desarrollo sexual 46,XY)
Pseudohermafroditismo femenino (Trastorno del desarrollo sexual 46, XX)